1 材料与方法
1.1 主要试剂
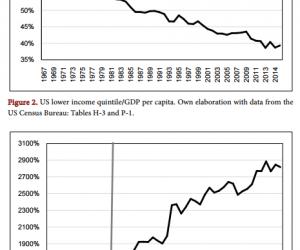
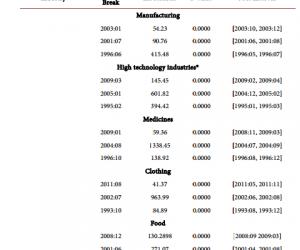
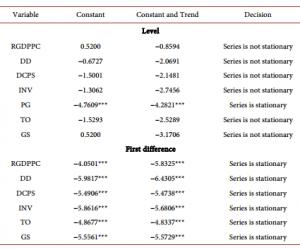
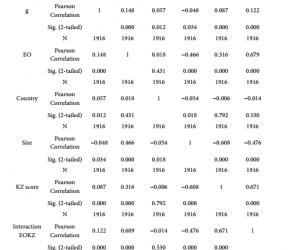
质粒提取试剂盒及胶回收试剂盒购自德国QIAGEN公司;细胞培养液DMEM购自美国Gibco公司;LipotectamineTM2000购自美国Invitrogen公司;PrP特异性单克隆抗体3F4购自丹麦Deko公司;6G3、β-actin和Bcl-2单克隆抗体及Dpl、Bax和casepase-3多克隆抗体均购自美国Santa Cruz公司;ECL Kit购自美国Perkin-Elmer公司;四甲基偶氮唑盐(MTT)购自美国Sigma公司;Annexin V/PI双染试剂盒购自上海宝赛生物公司。
1.2 真核表达质粒的构建
人全长PrP基因表达质粒pcDNA3.1-huPrP1-253及全长Dpl基因表达质粒pcDNA3.1-huDpl1-176由本室保存。pcDNA3.1-huPrPΔ32-121的PCR扩增:根据人PRNP基因序列设计引物,上游引入BamHⅠ,下游引入EcoRⅠ酶切位点(上游5′-GCGGATCCATGGCGAACCTTGGCTGCTG GATG-3′,下游5′-GCGAATTCTCAT-
CCCACTATCAGGAAGATGA-3′);另外一对中间引物(上游5′-CAGCATGTAGCCGCCAAGGCCCCCGTTCCATCCTCCAGGCTTCGGGCG-3′,下游5′-CGCCCGAAGCCTGGAGGATGGAACGGG-
GGCCTTGGCGGCTACATGCTG-3′)。PCR反应条件:94℃ 30s,63℃ 30s,72℃ 30s,共30个循环。PCR产物回收后与载体pMD18-T连接,经酶切后回收产物连接至pcDNA3.1(zeo+)真核表达载体,酶切及测序鉴定序列正确。
1.3 细胞培养及转染
SH-SY5Y细胞培养液为含4.5g/L谷氨酸和100mL/L胎牛血清的DMEM。细胞用6孔板/96孔板培养至对数期生长期,每孔转染的总DNA量分别为2μg/0.2μg,转染使用LipofectamineTM2000转染试剂盒。
1.4 Western blot
瞬时转染SH-SY5Y细胞24、48h后收获6孔板细胞,刮下的细胞用PBS洗涤2次,5000r/min离心5min;在细胞裂解缓冲液(5mL/L Triton X-100,5g/L去氧胆酸钠等)中4℃孵育30min。样品煮沸10min后进行150g/L SDS-PAGE并转移至硝酸纤维素膜上,一抗用含有50g/L脱脂奶粉的PBST(1∶600)稀释,4℃过夜;二抗用同上的PBST(1∶4000)稀释,最后用ECL显色试剂盒显色。
1.5 间接免疫荧光(IFA)
转染后24h的细胞爬片用40g/L多聚甲醛溶液固定,室温15min,2mL/L Triton-X100通透5min,含100mL/L胎牛血清的PBST封闭45min,加一抗37℃孵育2h,二抗(FITC标记)室温孵育1h,复染后显微镜下观察。
1.6 MTT检测
瞬时转染24、48h的96孔板细胞,每孔加入5mg/mL的MTT 20μL,继续培养4h,吸去培养液;加入200μL DMSO,摇床震荡10min,使结晶物充分溶解;450nm处用酶标仪测吸光度(A)值。实验重复3次,取其平均值。细胞存活率=实验组A值/对照组A值×100%。
1.7 流式细胞仪检测
瞬时转染24/48h的6孔板细胞,胰酶消化后用PBST洗涤2次,加入Annexin V和PI染料室温避光孵育15min,流式细胞仪上机检测。具体参见上海宝赛生物公司Annexin V/PI双染试剂盒说明书。
1.8 统计学处理
Western blot图像定量分析使用Total Tech图像处理软件(Pharmacia,美国)。数据以条形图表示,采用t检验进行统计学分析。每组数据的平均数均为进行3次独立实验的结果,并以平均数±标准差表示。以空质粒pcDNA3.1组作为对照组进行统计分析。P<0.05为差异有统计学意义。
2 结 果
2.1 Dpl及PrPs蛋白表达的鉴定
质粒pcDNA3.1-huPrP1-253、pcDNA3.1-huPrPΔ32-121和pcDNA3.1-huDpl1-176瞬时转染SH-SY5Y细胞24h后,收获细胞进行间接免疫荧光检测。结果可见,实验组的细胞膜上均出现了明显的绿色荧光,提示各重组表达质粒均在细胞中表达外源蛋白(图1)。
2.2 Dpl及PrPΔ32-121可诱导明显的细胞毒性效应
为检测瞬时转染SH-SY5Y细胞后表达的Dpl及PrPΔ32-121蛋白对细胞生长的影响,我们分别在转染了24、48h后进行MTT实验。结果发现,无论转染后24h还是48h,Dpl和PrPΔ32-121组较对照组(空载体及PrP1-253组)均出现了明显的细胞生存率下降现象,差异有统计学意义(P<0.05,图2)。
2.3 Dpl及PrPΔ32-121表达后凋亡相关蛋白的表达情况 国家级教育论文发表
为了观察Dpl及PrPΔ32-121蛋白表达后凋亡相关因子Bcl-2、Bax及caspase-3水平的变化情况,我们将重组质粒pcDNA3.1-huPrP1-253、pcDNA3.1-huPrPΔ32 -121和pcDNA3.1-huDpl1-176瞬时转染SH-SY5Y细胞48h后,提取细胞蛋白,进行Bcl-2、Bax及caspase-3(即全长无活性的pro-caspase-3)的Western blot检测。条带经凝胶灰度扫描软件分析后发现,与空载体对照组相比,Dpl及PrPΔ32-121实验组凋亡促进因子Bax蛋白的水平几乎没有变化,而凋亡抑制因子Bcl-2的水平出现了显著的下降(P<0.01,图3),caspase家族的pro-caspase-3蛋白由于被切割激活而显著降低(P<0.05,图3);而PrP1-253组则与空载体组几乎一致。
2.4 细胞凋亡情况
为进一步确证Dpl及PrPΔ32-121诱导细胞凋亡的效应,我们在pcDNA3.1-huPrP1-253、pcDNA3.1-huPrPΔ32-121和pcDNA3.1-huDpl1-176质粒转染SH-SY5Y细胞48h后,采用Annexin-V/PI双染流式细胞技术检测细胞凋亡情况。双染结果显示,Annexin-V/PI双染阳性细胞数在转染了pcDNA3.1、pcDNA3.1-huPrP1-253组中大致相同,无明显差异;而Dpl组(P<0.01)及PrPΔ32-121组(P<0.05)与对照组(pcDNA3.1组)相比,双染阳性细胞率均有明显的提高,并呈现统计学差异(图4)。
3 讨 论
作为一种朊蛋白样蛋白,Dpl蛋白与野生型全长PrP蛋白(由253个氨基酸组成,N端具有信号肽,C端有GPI锚)的构象相似,特别是与截短型的PrP突变体PrPΔ32-121更加相似,例如均有3个α-螺旋(αA、αB、αC)和2个反向平行的β-折叠。尽管Dpl和PrPΔ32-121在蛋白质一级和高级结构上也存在着差异,但通过我们的实验证实,其结构的相似性导致了二者的生物活性和细胞毒性作用的一致性。
通过MTT实验,证实Dpl蛋白和PrPΔ32-121能够引起相同的细胞毒性作用,而PrP1-253蛋白无此毒性作用,而且这种细胞毒性作用很可能是由细胞凋亡引起的。为此,我们进行了细胞凋亡相关指标的检测。结果显示,Dpl和PrPΔ32-121蛋白引起相似的细胞凋亡现象,包括无活性的pro-caspase-3水平的降低、Annexin V/PI阳性染色细胞数目增多和Bcl-2蛋白水平降低等。由Annexin V/PI实验和无活性pro-caspase-3蛋白水平的检测,我们推测Dpl和PrPΔ32-121蛋白的表达启动了细胞凋亡。Dpl和PrPΔ32-121蛋白的瞬时表达可能激活了无活性的pro-caspase-10或pro-caspase-9蛋白成为有活性的蛋白,继而级联地激活无活性的pro-caspase-3,被激活的caspase-3启动了细胞的凋亡过程。国家级教育论文发表
细胞凋亡一般由3条途径介导——线粒体途径、内质网途径和受体介导的凋亡。而Bcl-2是发生线粒体途径凋亡的关键指标,所以根据Bcl-2水平降低的结果,我们推测Dpl和PrPΔ32-121蛋白引起的细胞凋亡是由线粒体途径介导的。
以往研究显示,PrP基因缺失的小鼠其神经细胞要比野生型小鼠细胞对凋亡因素更加敏感[12-13],提示PrP蛋白具有细胞保护作用。大量研究也证明,8肽重复区是PrP神经保护性作用所必须的[8-9]。在PrP蛋白中缺失或插入额外的8肽序列都与人类的遗传型克雅氏病相关[10-11]。对于PrP蛋白分子来说,维持正确的8肽数目对于很多的生物功能是至关重要的,如对抗氧化应激的影响、与微管蛋白结合和抑制微管的组装等[9]。当然,我们不能否认除了8肽重复区的改变外,PrP的其他构象变化也可能影响到细胞的生长状态,但8肽重复区数量的准确和序列的完整是PrP蛋白的神经细胞保护效应的关键因素。








论文发表 是一个专门从事期刊推广、论文发表、论文发表辅导的机构。融合收集数百家期刊杂志社征稿评职称,发表论文,以供广大作者免费阅览,以期待各位作者在短时间内掌握每种期刊征稿要求、审稿范围,在日常繁重的工作中短时间开心、放心快速投稿、在适合的时间拿到刊物,评上职称,不为挤不上评职称的末班车而烦恼。
本站主要整合如下评职称,发表论文:
教育论文发表
经济论文发表
科技论文发表
医学论文发表
计算机论文发表
文学论文发表
农业论文发表
学报论文发表
其它论文发表等,另期待更多的杂志编辑与本站联系文:
85782530(普刊发表)
82534308(核心发表)共同以正期刊界正刊之气,维护学术良好的氛围。
免责声明:本网所提供的信息资源如有侵权、违规,请及时告知!
论文网版权所有, 本站提供论文发表 论文投稿 发表论文 发表文章
文章只代表作者观点,并不意味着本站认同,部分作品系转载,版权归原作者或相应的机构;若某篇作品侵犯您的权利,请来信告知:lunww@126.com
中国论文网全权所属国际域名:www.lunww.com 中文域名:论文发表 技术支持:论文发表 国家双软认定单位 法律顾问:全民安律师事务所
期刊合作加盟咨询:5715378(加盟、投诉、建议) 论文发表咨询电话:18262951856 论文发表投稿邮箱:lunww@126.com
Powered by 论文发表 © 2010-2020

苏ICP备19023845号-4