环氧合酶-2(cyclooxygenase-2, COX-2)是转化花生四烯酸(arachidonic acid, AA)为不稳定的中间产物PGG2的限速酶[1-2]。COX-2参与兴奋性谷氨酸能神经传递和长时程突触可塑性[3-5],并在神经源性炎症发生中起关键作用。前列腺素E2(prostagladin E2, PGE2)主要来源于COX-2氧化途径,研究表明PGE2是COX-2介导的突触效能变化的重要信使[3,6]。已经有4种PGE2的受体(PGE2 receptors, EPs)被鉴定和克隆,分别为EP1、EP2、EP3和EP4及EP3的多剪接异构体[5,7]。EPs属于7次跨膜结构的G蛋白藕联受体(G protein-coupled receptors, GPCRs),这些EP受体亚型呈现明显不同的信号转导途径和细胞反应。EP1的激活增加细胞内Ca2+水平,并与Gq-PLC-IP3和蛋白激酶C(protein kinase C, PKC)相藕联;EP3是与百日咳毒素敏感的Gi蛋白关联,导致cAMP生成减少,而EP2和EP4与Gs-cAMP-PKA途径相藕联引起cAMP增加[5,7]。研究表明PGE2在调制突触活动的效能是通过激活突触前的EP2起作用的[8-9],但EP2在突触传递中的作用还远未阐明。本研究拟探讨EP2在海马齿状回穿通纤维-颗粒细胞突触传递中的作用及其信号转导机制。
1 材料与方法
1.1 海马脑片准备
采用C57BL/6雄性小鼠,体重20~25g。海马脑片的制备如文献所述[3-4]。简言之,小鼠断头后将脑迅速置于0~4℃以950mL/L O2+50mL/L CO2混合气饱和的低Ca2+/高-Mg2+的切片用液(mmol/L):2.5 KCl,7.0 MgCl2,28.0 NaHCO3,1.25 NaH2PO4,0.5 CaCl2,7.0葡萄糖,3.0丙酮酸,1.0抗坏血酸,234蔗糖。脑片被切成350μm厚的薄片,将其转移到含被氧饱和的人工脑脊液(artificial cerebrospinal fluid, ACSF)的烧杯中进行孵育,ACSF的组成为(mmol/L):125.0 NaCl, 2.5 KCl,1.0 MgCl2,25.0 NaHCO3,1.25 NaH2PO4,2.0 CaCl2,25.0葡萄糖,3.0丙酮酸,1.0抗坏血酸。在36℃孵育0.5~1h,然后置于室温(22~24℃)至少1.5h后可进行电生理记录。记录时将脑片转移至标本室,并在32~34℃连续地被950mL/L O2,50mL/L CO2饱和的ACSF灌流。在配有×60的入水镜头和直立式微分干涉相差显微镜 (Olympus BX51WI)下观察齿状回的颗粒细胞。随机抽取的每只小鼠制备的海马脑片取1~2片用于每组实验。用于每组实验的脑片为8~13片,对应的小鼠的数量是5~8只,共计使用小鼠44只用于实验。
1.2 主要试剂
Butaprost(Buta)、AH6809(AH)、Forskolin(Forsk)、H89、PD98059(PD)及2-APB均购自Sigma。上述药物分别溶解于二甲亚砜(dimethylsulfoxide,DMSO;Sigma),在临用前用ACSF分别稀释成所需浓度的工作液,工作液中DMSO的终浓度不超过1.4mmol/L。
1.3 电生理记录
使用Axoclamp-2B膜片钳放大器的桥式整流模式(bridge mode)在齿状回刺激穿通纤维,刺激频率0.05Hz,记录场兴奋性突触后电流(field excitatory post-synaptic potential, fEPSP)[4]。细胞外液组成为(mmol/L):130.0 NaCl,2.5 KCl,1.0 MgCl2,10.0 HEPES,1.25 NaH2PO4,2.0 CaCl2,25.0葡萄糖(pH 7.4)。记录电极充灌ACSF(2~4MΩ),置于齿状回的分子细胞层。取三分之一的fEPSP最大反应的刺激作为基线的刺激强度,双脉冲刺激(paired-pulse stimulation)诱导的脉冲间隔为40ms。双脉冲比值(paired-pulse ratio, PPR)是第2个EPSP的幅值(P2)与第1个EPSP的幅值(P1)之比,即P2/P1。所有的浴槽灌流液均含有荷包牡丹碱(bicuculine, 10μmol/L)以阻断GABA受体介导的反应。
1.4 统计学处理
所有数据以均数±标准误表示,均数间差异的显著性检验用Student?s t
-test及方差分析(ANOVA),在需要时可结合学生Newman-Keuls检验进行统计学差异的比较。P<0.05为差异有显著性。
2 结 果
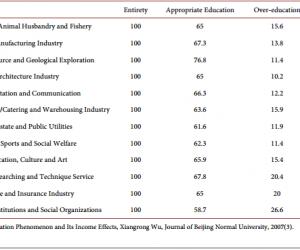
2.1 EP2的激动剂Buta增强海马齿状回突触传递职称论文发表费用
为阐明EP2在海马齿状回突触传递中的作用,观察EP2激动剂Buta对fEPSP的影响。记录fEPSP至少10min作为基线,给予Buta(5μmol/L)作用10min,然后用细胞外液冲洗30min至实验结束。Buta明显降低PPR,由用药前的1.32 ±0.054降低至用药后的1.09±0.035(6~10min)(P<0.01,图1A和1B);而在AH存在的情况下,Buta在用药前后PPR没有明显变化[(1.29±0.052)% vs. (1.25±0.048)%;P>0.05;图1A和1B]。Buta显著增强fEPSP的斜率[(153.92±5.72)%; n=13;P<0.01] (图1C),且这一效应被EP2的拮抗剂AH(10μmol/L)所阻断[(100.80±3.08)%;n=12;图1C和1D] 。上述结果表明EP2增强突触传递的效应是通过突触前机制起作用的。
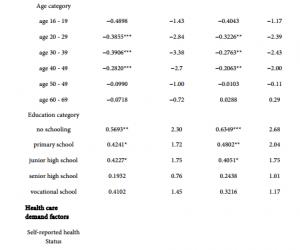
2.2 EP2拮抗剂AH对EP2的下游信号途径的影响
由于EP2与Gs-cAMP信号途径相藕联[5,8],因此使用Forsk激活腺苷酸环化酶,观察Forsk单独或Forsk+AH对fEPSP的影响。灌流Forsk(20μmol/L)引起fEPSP的斜率增加到(206.47±16.02)%(n=10,图2A和2B),而在AH(10μmol/L)存在的条件下,同样浓度的Forsk仍然可增强fEPSP的斜率到(217.23±20.38)%(n=8,图2B和2C)。结果提示cAMP是EP2作用的下游位点。
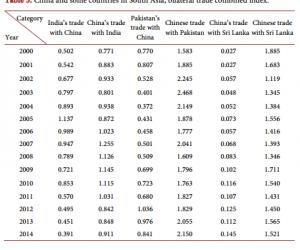
2.3 EP2增强突触效能的效应由PKA、ERK和IP3途径介导
由于EP2与Gs-cAMP/PKA途径相藕联,因此,以浴液灌流PKA的抑制剂H89观察其能否阻断EP2的激动剂Buta的效应。在H89(5μmol/L)存在的情况下,Buta(5μmol/L)增强突触效能的效应被显著抑制[Buta:(153.92±5.72)%, n=13;H89+Buta:(111.43±4.12)%, n=10; P<0.05](图3A)。
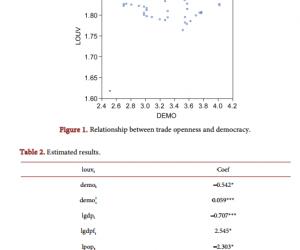
当分别给予ERK和IP3的抑制剂PD和2-APB,以确定这些信号转导途径是否参与了Buta介导的效应。如图3B、3C和3D所示,Buta(5μmol/L)所诱导的fEPSP斜率的增加分别被PD(20μmol/L)和2-APB(20μmol/L)完全阻断。结果表明ERK和IP3信号途径参与了Buta诱导的效应。
3 讨 论
COX-2是催化AA转化为PGs的限速酶[1],而PGE2是来源于COX-2氧化代谢途径的主要产物[5-6,8]。大量的研究表明COX-2和PGE2参与突触传递和可塑性,COX-2调制海马的突触传递和可塑性是由PGE2介导的[3,6]。GPCRs的EP1-4在海马和皮层的神经元呈异源性表达[10],且呈现明显不同的信号转导途径和细胞反应。EP1的激活增加细胞内Ca2+水平,并与Gq-PLC-IP3和蛋白激酶C (protein kinase C, PKC)相藕联;EP3与百日咳毒素敏感的Gi蛋白关联,导致cAMP的生成减少,而EP2和EP4与Gs-cAMP-PKA途径相藕联引起cAMP增加[5,7]。本研究的结果表明EP2受体的激活可增强突触传递,并且是通过突触前机制起作用。这与前人的研究PGE2在调制突触活动的效能是通过激活突触前的EP2起作用的观点相一致[8]。在视皮层抑制EP2基因可消除LTP的诱导[9]。职称论文发表费用
Forsk诱导的细胞内cAMP生成增加,无论有无EP2拮抗剂的存在,forsk均可增强突触传递,证明cAMP途径是EP2作用的下游位点。本研究也证实EP2的激动剂诱导的突触效能的增强是由PKA途径所介导。上述结果表明在海马齿状回EP2是通过cAMP/PKA途径起作用的。
细胞外信号调节蛋白激酶(extracellular signal-regulated protein kinase, ERK),p38 MAPK等与COX-2的表达和PGE2的产生有关。然而,关于EP2诱导的突触效能的变化是由哪些信号转导途径所介导还知之甚少。本研究表明ERK及IP3信号转导途径参与了EP2介导的突触传递的增强。这说明多个信号途径介导了EP2诱导的突触效应。








论文发表 是一个专门从事期刊推广、论文发表、论文发表辅导的机构。融合收集数百家期刊杂志社征稿评职称,发表论文,以供广大作者免费阅览,以期待各位作者在短时间内掌握每种期刊征稿要求、审稿范围,在日常繁重的工作中短时间开心、放心快速投稿、在适合的时间拿到刊物,评上职称,不为挤不上评职称的末班车而烦恼。
本站主要整合如下评职称,发表论文:
教育论文发表
经济论文发表
科技论文发表
医学论文发表
计算机论文发表
文学论文发表
农业论文发表
学报论文发表
其它论文发表等,另期待更多的杂志编辑与本站联系文:
85782530(普刊发表)
82534308(核心发表)共同以正期刊界正刊之气,维护学术良好的氛围。
免责声明:本网所提供的信息资源如有侵权、违规,请及时告知!
论文网版权所有, 本站提供论文发表 论文投稿 发表论文 发表文章
文章只代表作者观点,并不意味着本站认同,部分作品系转载,版权归原作者或相应的机构;若某篇作品侵犯您的权利,请来信告知:lunww@126.com
中国论文网全权所属国际域名:www.lunww.com 中文域名:论文发表 技术支持:论文发表 国家双软认定单位 法律顾问:全民安律师事务所
期刊合作加盟咨询:5715378(加盟、投诉、建议) 论文发表咨询电话:18262951856 论文发表投稿邮箱:lunww@126.com
Powered by 论文发表 © 2010-2020

苏ICP备19023845号-4