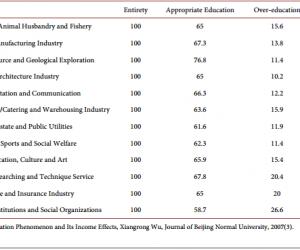
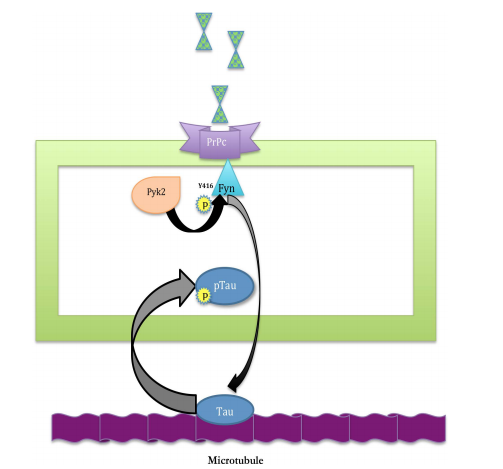
 Mapping It Out: A Novel Signaling Pathway Linking Aβ-PrPc-Fyn Complex to Cognitive Impairment in Alzheimer’s Disease Yiling Wang1*, Lei Xue2* 1 Department of Neuroscience, Duke University, Durham, USA 2 Shanghai Key Laboratory of Signaling and Disease Research, School of Life Science, Shanghai, China Email: * yiling.wang@duke.edu, * lei.xue@tongji.edu.cn Received 5 November 2013; revised 14 December 2013; accepted 21 December 2013 Copyright © 2014 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Understanding the signaling cascade that leads to the rapid memory and cognitive breakdown in Alzheimer’s disease is the key to finding a potential treatment method for the disease. Recently, Larson et al. connect the roles of major proteins implicated in the disease progression and propose targeting of cellular prion protein (PrPc) as a way of someday rescuing synaptic plasticity in humans with Alzheimer’s, as it has been done on mice. Keywords Alzheimer’s; PrPc; Aβ; Fyn; Tau; Microtubule 1. Introduction Alzheimer’s disease (AD), an often times-fatal form of dementia, is among the most prevalent neurodegenerative disorders alongside Parkinson’s disease and Amyotrophic Lateral Sclerosis. Two of the most prominent characteristics that define Alzheimer’s disease are amyloid plaques, which are solid deposits of beta-amyloid (Aβ) protein, and neurofibrillary tangles, comprised of insoluble aggregates of tau protein along with collapsed microtubules. For the past few decades, many have sought to understand the biological mechanisms underlying Alzheimer’s disease, and although much research has gone into deciphering the roles of the key players at work, up until recently, a detailed molecular pathway leading to Alzheimer’s disease had not yet been mapped out. Some of these key players in AD include Aβ oligomers, cellular prion protein (PrPc ), tau protein, and Fyn kinase. Working in conjunction with cellular prion protein (PrPc ), Aβ dimmers are largely responsible for inhibit- * Corresponding authors. Y. L. Wang, L. Xue 21 ing long-term potentiation (LTP) and inducing dendritic spine loss in hippocampal neurons [1], thus causing synaptic plasticity impairment in AD patients [2]. In other AD studies, hyperphosphorylation of tau (a microtubule-associated protein) caused tau mislocalization in dendritic spines, which disrupts synaptic plasticity and cognitive function [3]. Furthermore, cognitive impairment is not only associated with heightened levels of phosphorylated tau, but it is also linked to overexpression of the protein Fyn [4], a postsynaptic protein kinase implicated in the integrin signaling pathway [5]. Therefore, evidence from previous studies suggests that Aβ, PrPc , tau, and Fyn somehow all contribute to the synaptic impairment and cognitive decline in AD patients. However, the link between the Aβ-PrPc -Fyn complex and tau hyperphosphorylation and how it leads to dementia in AD remains unclear. 2. Role of PrPc-Dimeric Aβ Signaling A primary goal of this paper by Larson et al. is to elucidate the molecular mechanism (Figure 1) by which oli gomeric Aβ, coupled with PrPc receptor, phosphorylates the tyrosine kinase Fyn and produces neurofibrillary tangles in AD [5]. Through confocal immunofluorescence, Larson et al. observed that colocalization of PrPc and Aβ occurred predominantly on synaptic sites, providing evidence that PrPc serves as a membrane receptor for Figure 1. A schematic of the signaling cascade described by Larson et al. (2012), starting with Aβ dimer- PrPc binding to the disruption of tau function. (From top to bottom): (I) Endogenous Aβ dimers bind to PrPc receptors on cell surface; (II) Once Aβ binds PrPc , a signaling cascade is triggered in which Pyk2 (an integrin subunit) phosphorylates Fyn at the Y416 site (a tyrosine protein kinase to which PrPc is coupled) and activates Fyn; (III) Phosphorylated Fyn moves to the dendritic spine where microtubules are mediated by the protein tau; (IV) Fyn acts on tau to hyperphosphorylate it, leading to tau dysfunction, microtubule destabilization, and impairment of synaptic plasticity. Y. L. Wang, L. Xue 22 the Aβ ligand (Figure 1(I)). Ultimately, Larson et al. characterize a signaling cascade in AD, through which dimeric Aβ acts as a ligand for the PrPc receptor and activates intracellular Fyn through phosphorylation (Figure 1(II)). Fyn activation in turn hyperphosphorylates and mislocalizes tau protein in the dendritic spines, leading to destabilized microtubules, which produce neurofibrillary tangles and the cognitive impairment characteristic of AD patients (Figures 1(III) and (IV)). The authors found that AD brain tissue contained greater levels of cellular prion protein (PrPc ) expression, which was positively correlated with Fyn phosphorylation. These observations support previous studies pointing to Fyn’s role in mediating cognitive defects associated with AD [5]. But here, Larson et al. draws a novel connection between PrPc and Fyn, suggesting that their interaction in conjunction with the presence of Aβ oligomers may be bringing about the neurological impairments in AD. 3. Discussion However, in opposition to Larson et al.’s findings asserting the necessity of PrPc in mediating synaptic toxicity, several other studies produced results that are not consistent to this claim. Notably, in two studies done in 2010, APP-expressing transgenic mice displayed no significant difference in cognitive performance regardless of PrPc overexpression or ablation [6] [7]. These studies that failed to show synaptic impairment in AD as dependent upon PrPc overexpression [7] stand in direct opposition to Larson et al.’s findings. Yet, in defense of Larson et al.’s conclusion, we must consider that, whereas the research conflicting with Larson et al.’s results experimented with 2-to-4-month-old Prnp transgenic mice, the age at which amyloid plaques actually begin forming in mice brains is no earlier than 10 - 14 months old [5]. Since these beta-amyloid plaques found only in older mice comprise Aβ dimers, and since Aβ dimers are the only beta-amyloid species to bind PrPc receptors, only in the AD mice older than 10 months can we see the effect that PrPc receptor exerts on synaptic plasticity impairment once it binds dimeric Aβ [5]. Therefore, these studies [6] [7] failed to observe PrPc ’s effects on LTP impairment as reported by Larson et al., not because PrPc is not implicated in the signaling pathway, but rather because the mice they experimented with were too young to have actually developed the Aβ dimers that bind PrPc to initiate the signaling cascade. 3.1. Significance of Tau Mislocalization in Alzheimer’s Disease The paper by Larson et al. is essentially the first to characterize the signaling cascade linking endogenous Aβ dimer with the mislocalization and hyperphosphorylation of tau in Alzheimer’s disease [5]. It consolidates evidence from previous papers illustrating the roles that various molecules play to produce neuronal dysfunction in AD. This paper pieces together the bits of information gathered from research over the decades so that this proposed pathway has logical flow throughout. Previous studies have documented the consequences of PrPc and Aβ interaction on LTP impairment [1], reversal of synaptotoxic effects by Aβ due to genetic ablation of Fyn [4], and neurofibrillary tangles resulting from tau hyperphosphorylation [3]. But prior to this paper, it was unclear how these phenomena were related to produce Alzheimer’s disease. Therefore, Larson et al. were the first to draw a link among these findings and prove a logical signaling cascade in which Aβ dimer binds to the PrPc receptor to activate Fyn, which then goes on to phosphorylate tau and cause tau missorting in the dendritic spine, and this tau mislocalization eventually leads to synaptic plasticity inhibition. 3.2. Potential Treatment Methods Targeting PrPc-Aβ Dimer Binding In addition, Larson et al. cites PrPc targeting as a potentially viable method for slowing down the progression of Alzheimer’s disease in humans, since blocking PrPc -Aβ dimer binding using the antibody 6D11 in AD mice resulted in the reversal of LTP impairment [5], or LTP rescue. Furthermore, the authors add that since PrPc receptors located on the cell membranes are readily accessible, targeting them as a possible treatment method is entirely feasible to do in the near future. Finally, this paper allows us to see why neurofibrillary tangles and amyloid-beta plaques appear simultaneously in AD brain tissue: the Aβ dimers that comprise plaques directly influence phoshporylated tau aggregation into neurofibrillary tangles. 3.3. Future Directions To build upon the knowledge that Larson et al. have presented, future research may strive to find a safe and ef- Y. L. Wang, L. Xue 23 fective way to block PrPc -Aβ dimer binding in humans so as to potentially rescue LTP in Alzheimer’s patients. Furthermore, one may try to find any link between the integrin signaling cascade and the proposed pathway, both of which involve the Fyn kinase, as such knowledge may allow us to gain a broader perspective of how interconnected the various molecules and pathways are in the AD neuron. Acknowledgements This study was supported by Shanghai Committee of Science and Technology (grant No. 09DZ2260100). References [1] Shankar, G.M., Li, S., Mehta, T.H., Garcia-Munoz, A., Shepardson, N.E., Smith, I., Brett, F.M., Farrell, M.A., Rowan, M.J., Lemere, C.A., Regan, C.M., Walsh, D.M., Sabatini, B.L., and Selkoe, D.J. (2008) Amyloid β-Protein Dimers Isolated Directly from Alzheimer Brains Impair Synaptic Plasticity and Memory. Nature Medicine, 8, 837-842. http://dx.doi.org/10.1038/nm1782 [2] Laurén, J., Gimbel, D.A., Nygaard, H., Gilbert, J.W. and Strittmatter, S.M. (2009) Cellular Prion Protein Mediates Impairment of Synaptic Plasticity by Amyloid-Beta Oligomers. Nature, 457, 1128-1132. http://dx.doi.org/10.1038/nature07761 [3] Hoover, B.R., Reed, M.N., Su, J., Penrod, R.D., Kotilinek, L.A., Grant, M.K., Pitstick, R., Carlson, G.A., Lanier, L.M., Yuan, L.L., Ashe, K.H. and Liao, D. (2010) Tau Mislocalization to Dendritic Spines Mediates Synaptic Dysfunction Independently of Neurodegeneration. Neuron, 68, 1067-1081. http://dx.doi.org/10.1016/j.neuron.2010.11.030 [4] Roberson, E.D., Halabisky, B., Yoo, J.W., Yao, J., Chin, J., Yan, F., Wu, T., Hamto, P., Devidze, N., Yu, G.Q., Palop, J.J., Noebels, J.L. and Mucke, L. (2011) Amyloid-Beta/Fyn-Induced Synaptic, Network, and Cognitive Impairments Depend on Tau Levels in Multiple Mouse Models of Alzheimer’s Disease. The Journal of Neuroscience, 31, 700 -711. http://dx.doi.org/10.1523/JNEUROSCI.4152-10.2011 [5] Larson, M., Sherman, M.A., Amar, F., Nuvolone, M., Schneider, J.A., Bennett, D.A., Aguzzi, A. and Lesné, S.E. (2012) The Complex PrPc-Fyn Couples Human Oligomeric Aβ with Pathological Tau Changes in Alzheimer’s Disease. The Journal of Neuroscience, 32, 16857-16871. http://dx.doi.org/10.1523/JNEUROSCI.1858-12.2012 [6] Calella, A.M., Farinelli, M., Nuvolone, M., Mirante, O., Moos, R., Falsig, J., Mansuy, I.M. and Aguzzi, A. (2010) Prion Protein and Abeta-Related Synaptic Toxicity Impairment. EMBO Molecular Medicine, 2, 306 -314. http://dx.doi.org/10.1002/emmm.201000082 [7] Balducci, C., Beeg, M., Stravalaci, M., Bastone, A., Sclip, A., Biasini, E., Tapella, L., Colombo, L., Manzoni, C., Borsello, T., Chiesa, R., Gobbi, M., Salmona, M. and Forloni, G. (2010) Synthetic Amyloid-β Oligomers Impair LongTerm Memory Independently of Cellular Prion Protein. Proceedings of the National Academy of Sciences, 107, 2295- 2300. http://dx.doi.org/10.1073/pnas.0911829107
Mapping It Out: A Novel Signaling Pathway Linking Aβ-PrPc-Fyn Complex to Cognitive Impairment in Alzheimer’s Disease Yiling Wang1*, Lei Xue2* 1 Department of Neuroscience, Duke University, Durham, USA 2 Shanghai Key Laboratory of Signaling and Disease Research, School of Life Science, Shanghai, China Email: * yiling.wang@duke.edu, * lei.xue@tongji.edu.cn Received 5 November 2013; revised 14 December 2013; accepted 21 December 2013 Copyright © 2014 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Understanding the signaling cascade that leads to the rapid memory and cognitive breakdown in Alzheimer’s disease is the key to finding a potential treatment method for the disease. Recently, Larson et al. connect the roles of major proteins implicated in the disease progression and propose targeting of cellular prion protein (PrPc) as a way of someday rescuing synaptic plasticity in humans with Alzheimer’s, as it has been done on mice. Keywords Alzheimer’s; PrPc; Aβ; Fyn; Tau; Microtubule 1. Introduction Alzheimer’s disease (AD), an often times-fatal form of dementia, is among the most prevalent neurodegenerative disorders alongside Parkinson’s disease and Amyotrophic Lateral Sclerosis. Two of the most prominent characteristics that define Alzheimer’s disease are amyloid plaques, which are solid deposits of beta-amyloid (Aβ) protein, and neurofibrillary tangles, comprised of insoluble aggregates of tau protein along with collapsed microtubules. For the past few decades, many have sought to understand the biological mechanisms underlying Alzheimer’s disease, and although much research has gone into deciphering the roles of the key players at work, up until recently, a detailed molecular pathway leading to Alzheimer’s disease had not yet been mapped out. Some of these key players in AD include Aβ oligomers, cellular prion protein (PrPc ), tau protein, and Fyn kinase. Working in conjunction with cellular prion protein (PrPc ), Aβ dimmers are largely responsible for inhibit- * Corresponding authors. Y. L. Wang, L. Xue 21 ing long-term potentiation (LTP) and inducing dendritic spine loss in hippocampal neurons [1], thus causing synaptic plasticity impairment in AD patients [2]. In other AD studies, hyperphosphorylation of tau (a microtubule-associated protein) caused tau mislocalization in dendritic spines, which disrupts synaptic plasticity and cognitive function [3]. Furthermore, cognitive impairment is not only associated with heightened levels of phosphorylated tau, but it is also linked to overexpression of the protein Fyn [4], a postsynaptic protein kinase implicated in the integrin signaling pathway [5]. Therefore, evidence from previous studies suggests that Aβ, PrPc , tau, and Fyn somehow all contribute to the synaptic impairment and cognitive decline in AD patients. However, the link between the Aβ-PrPc -Fyn complex and tau hyperphosphorylation and how it leads to dementia in AD remains unclear. 2. Role of PrPc-Dimeric Aβ Signaling A primary goal of this paper by Larson et al. is to elucidate the molecular mechanism (Figure 1) by which oli gomeric Aβ, coupled with PrPc receptor, phosphorylates the tyrosine kinase Fyn and produces neurofibrillary tangles in AD [5]. Through confocal immunofluorescence, Larson et al. observed that colocalization of PrPc and Aβ occurred predominantly on synaptic sites, providing evidence that PrPc serves as a membrane receptor for Figure 1. A schematic of the signaling cascade described by Larson et al. (2012), starting with Aβ dimer- PrPc binding to the disruption of tau function. (From top to bottom): (I) Endogenous Aβ dimers bind to PrPc receptors on cell surface; (II) Once Aβ binds PrPc , a signaling cascade is triggered in which Pyk2 (an integrin subunit) phosphorylates Fyn at the Y416 site (a tyrosine protein kinase to which PrPc is coupled) and activates Fyn; (III) Phosphorylated Fyn moves to the dendritic spine where microtubules are mediated by the protein tau; (IV) Fyn acts on tau to hyperphosphorylate it, leading to tau dysfunction, microtubule destabilization, and impairment of synaptic plasticity. Y. L. Wang, L. Xue 22 the Aβ ligand (Figure 1(I)). Ultimately, Larson et al. characterize a signaling cascade in AD, through which dimeric Aβ acts as a ligand for the PrPc receptor and activates intracellular Fyn through phosphorylation (Figure 1(II)). Fyn activation in turn hyperphosphorylates and mislocalizes tau protein in the dendritic spines, leading to destabilized microtubules, which produce neurofibrillary tangles and the cognitive impairment characteristic of AD patients (Figures 1(III) and (IV)). The authors found that AD brain tissue contained greater levels of cellular prion protein (PrPc ) expression, which was positively correlated with Fyn phosphorylation. These observations support previous studies pointing to Fyn’s role in mediating cognitive defects associated with AD [5]. But here, Larson et al. draws a novel connection between PrPc and Fyn, suggesting that their interaction in conjunction with the presence of Aβ oligomers may be bringing about the neurological impairments in AD. 3. Discussion However, in opposition to Larson et al.’s findings asserting the necessity of PrPc in mediating synaptic toxicity, several other studies produced results that are not consistent to this claim. Notably, in two studies done in 2010, APP-expressing transgenic mice displayed no significant difference in cognitive performance regardless of PrPc overexpression or ablation [6] [7]. These studies that failed to show synaptic impairment in AD as dependent upon PrPc overexpression [7] stand in direct opposition to Larson et al.’s findings. Yet, in defense of Larson et al.’s conclusion, we must consider that, whereas the research conflicting with Larson et al.’s results experimented with 2-to-4-month-old Prnp transgenic mice, the age at which amyloid plaques actually begin forming in mice brains is no earlier than 10 - 14 months old [5]. Since these beta-amyloid plaques found only in older mice comprise Aβ dimers, and since Aβ dimers are the only beta-amyloid species to bind PrPc receptors, only in the AD mice older than 10 months can we see the effect that PrPc receptor exerts on synaptic plasticity impairment once it binds dimeric Aβ [5]. Therefore, these studies [6] [7] failed to observe PrPc ’s effects on LTP impairment as reported by Larson et al., not because PrPc is not implicated in the signaling pathway, but rather because the mice they experimented with were too young to have actually developed the Aβ dimers that bind PrPc to initiate the signaling cascade. 3.1. Significance of Tau Mislocalization in Alzheimer’s Disease The paper by Larson et al. is essentially the first to characterize the signaling cascade linking endogenous Aβ dimer with the mislocalization and hyperphosphorylation of tau in Alzheimer’s disease [5]. It consolidates evidence from previous papers illustrating the roles that various molecules play to produce neuronal dysfunction in AD. This paper pieces together the bits of information gathered from research over the decades so that this proposed pathway has logical flow throughout. Previous studies have documented the consequences of PrPc and Aβ interaction on LTP impairment [1], reversal of synaptotoxic effects by Aβ due to genetic ablation of Fyn [4], and neurofibrillary tangles resulting from tau hyperphosphorylation [3]. But prior to this paper, it was unclear how these phenomena were related to produce Alzheimer’s disease. Therefore, Larson et al. were the first to draw a link among these findings and prove a logical signaling cascade in which Aβ dimer binds to the PrPc receptor to activate Fyn, which then goes on to phosphorylate tau and cause tau missorting in the dendritic spine, and this tau mislocalization eventually leads to synaptic plasticity inhibition. 3.2. Potential Treatment Methods Targeting PrPc-Aβ Dimer Binding In addition, Larson et al. cites PrPc targeting as a potentially viable method for slowing down the progression of Alzheimer’s disease in humans, since blocking PrPc -Aβ dimer binding using the antibody 6D11 in AD mice resulted in the reversal of LTP impairment [5], or LTP rescue. Furthermore, the authors add that since PrPc receptors located on the cell membranes are readily accessible, targeting them as a possible treatment method is entirely feasible to do in the near future. Finally, this paper allows us to see why neurofibrillary tangles and amyloid-beta plaques appear simultaneously in AD brain tissue: the Aβ dimers that comprise plaques directly influence phoshporylated tau aggregation into neurofibrillary tangles. 3.3. Future Directions To build upon the knowledge that Larson et al. have presented, future research may strive to find a safe and ef- Y. L. Wang, L. Xue 23 fective way to block PrPc -Aβ dimer binding in humans so as to potentially rescue LTP in Alzheimer’s patients. Furthermore, one may try to find any link between the integrin signaling cascade and the proposed pathway, both of which involve the Fyn kinase, as such knowledge may allow us to gain a broader perspective of how interconnected the various molecules and pathways are in the AD neuron. Acknowledgements This study was supported by Shanghai Committee of Science and Technology (grant No. 09DZ2260100). References [1] Shankar, G.M., Li, S., Mehta, T.H., Garcia-Munoz, A., Shepardson, N.E., Smith, I., Brett, F.M., Farrell, M.A., Rowan, M.J., Lemere, C.A., Regan, C.M., Walsh, D.M., Sabatini, B.L., and Selkoe, D.J. (2008) Amyloid β-Protein Dimers Isolated Directly from Alzheimer Brains Impair Synaptic Plasticity and Memory. Nature Medicine, 8, 837-842. http://dx.doi.org/10.1038/nm1782 [2] Laurén, J., Gimbel, D.A., Nygaard, H., Gilbert, J.W. and Strittmatter, S.M. (2009) Cellular Prion Protein Mediates Impairment of Synaptic Plasticity by Amyloid-Beta Oligomers. Nature, 457, 1128-1132. http://dx.doi.org/10.1038/nature07761 [3] Hoover, B.R., Reed, M.N., Su, J., Penrod, R.D., Kotilinek, L.A., Grant, M.K., Pitstick, R., Carlson, G.A., Lanier, L.M., Yuan, L.L., Ashe, K.H. and Liao, D. (2010) Tau Mislocalization to Dendritic Spines Mediates Synaptic Dysfunction Independently of Neurodegeneration. Neuron, 68, 1067-1081. http://dx.doi.org/10.1016/j.neuron.2010.11.030 [4] Roberson, E.D., Halabisky, B., Yoo, J.W., Yao, J., Chin, J., Yan, F., Wu, T., Hamto, P., Devidze, N., Yu, G.Q., Palop, J.J., Noebels, J.L. and Mucke, L. (2011) Amyloid-Beta/Fyn-Induced Synaptic, Network, and Cognitive Impairments Depend on Tau Levels in Multiple Mouse Models of Alzheimer’s Disease. The Journal of Neuroscience, 31, 700 -711. http://dx.doi.org/10.1523/JNEUROSCI.4152-10.2011 [5] Larson, M., Sherman, M.A., Amar, F., Nuvolone, M., Schneider, J.A., Bennett, D.A., Aguzzi, A. and Lesné, S.E. (2012) The Complex PrPc-Fyn Couples Human Oligomeric Aβ with Pathological Tau Changes in Alzheimer’s Disease. The Journal of Neuroscience, 32, 16857-16871. http://dx.doi.org/10.1523/JNEUROSCI.1858-12.2012 [6] Calella, A.M., Farinelli, M., Nuvolone, M., Mirante, O., Moos, R., Falsig, J., Mansuy, I.M. and Aguzzi, A. (2010) Prion Protein and Abeta-Related Synaptic Toxicity Impairment. EMBO Molecular Medicine, 2, 306 -314. http://dx.doi.org/10.1002/emmm.201000082 [7] Balducci, C., Beeg, M., Stravalaci, M., Bastone, A., Sclip, A., Biasini, E., Tapella, L., Colombo, L., Manzoni, C., Borsello, T., Chiesa, R., Gobbi, M., Salmona, M. and Forloni, G. (2010) Synthetic Amyloid-β Oligomers Impair LongTerm Memory Independently of Cellular Prion Protein. Proceedings of the National Academy of Sciences, 107, 2295- 2300. http://dx.doi.org/10.1073/pnas.0911829107







论文发表 是一个专门从事期刊推广、论文发表、论文发表辅导的机构。融合收集数百家期刊杂志社征稿评职称,发表论文,以供广大作者免费阅览,以期待各位作者在短时间内掌握每种期刊征稿要求、审稿范围,在日常繁重的工作中短时间开心、放心快速投稿、在适合的时间拿到刊物,评上职称,不为挤不上评职称的末班车而烦恼。
本站主要整合如下评职称,发表论文:
教育论文发表
经济论文发表
科技论文发表
医学论文发表
计算机论文发表
文学论文发表
农业论文发表
学报论文发表
其它论文发表等,另期待更多的杂志编辑与本站联系文:
85782530(普刊发表)
82534308(核心发表)共同以正期刊界正刊之气,维护学术良好的氛围。
免责声明:本网所提供的信息资源如有侵权、违规,请及时告知!
论文网版权所有, 本站提供论文发表 论文投稿 发表论文 发表文章
文章只代表作者观点,并不意味着本站认同,部分作品系转载,版权归原作者或相应的机构;若某篇作品侵犯您的权利,请来信告知:lunww@126.com
中国论文网全权所属国际域名:www.lunww.com 中文域名:论文发表 技术支持:论文发表 国家双软认定单位 法律顾问:全民安律师事务所
期刊合作加盟咨询:5715378(加盟、投诉、建议) 论文发表咨询电话:18262951856 论文发表投稿邮箱:lunww@126.com
Powered by 论文发表 © 2010-2020

苏ICP备19023845号-4